Fibrosi cistica
Che cosa è la fibrosi cistica?
La fibrosi cistica è una malattia ereditaria a carico delle ghiandole secretorie, tra cui quelle che producono muco e le ghiandole sudoripare.
La fibrosi cistica si caratterizza per la produzione di muco che ostruisce le cavità di molti organi cavi.
Gli organi più compromessi in questa malattia sono polmoni, fegato, pancreas, intestino e organi sessuali.
Essa colpisce uomini e donne di tutti i gruppi etnici. Sono circa 70000 le persone affette in tutto il mondo e ogni anno si aggiungono 1000 nuovi casi.
Il numero maggiore di malati di fibrosi cistica si registra tra i discendenti della popolazione caucasica del nord Europa: la frequenza di nascita di un bambino affetto è di 1 su 2000-3000. Negli Stati Uniti si riscontra un'incidenza simile. La malattia è diffusa anche tra i Latini e gli Indiani d'America. Le frequenze più basse (1 su 30000) si riscontrano tra gli Americani di origine asiatica e africana.
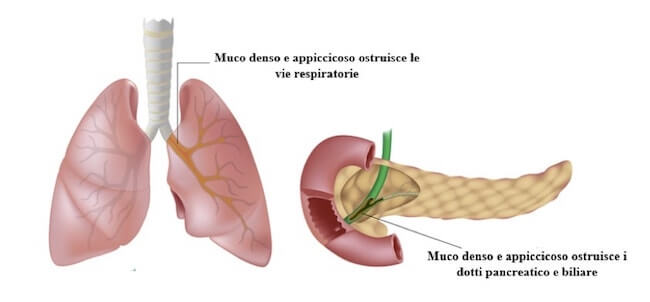
La manifestazione tipica della fibrosi cistica è la produzione di muco denso. I sistemi digerente e respiratorio sono i più compromessi e la morte sopraggiunge spesso per complicazioni a carico degli organi che ne fanno parte.
Segni e sintomi a carico dell'apparato respiratorio
La manifestazione tipica della fibrosi cistica è la produzione di muco denso e appiccicoso che può ostruire le vie respiratorie e provocare difficoltà respiratorie e infezioni batteriche che causano tosse e infiammazione continui. I violenti colpi di tosse producono espettorati che possono recare tracce di sangue.
La presenza di muco favorisce l'attecchimento di batteri del genere Pseudomonas, responsabili di infezioni spesso non trattabili per via della resistenza a molti antibiotici.
L'ampia infiltrazione di neutrofili è inoltre responsabile, insieme alla presenza di muco, della distruzione e della cicatrizzazione del tessuto polmonare e della formazione di cisti. Ulteriori complicazioni riguardano il rischio continuo di bronchiti e polmoniti e di serie problematiche come pneumotorace e bronchiectasia.
In alcuni pazienti possono formarsi dei polipi nasali che richiedono l'intervento chirurgico.
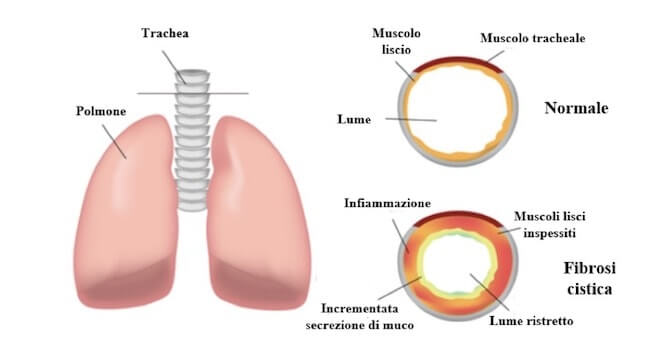
Il muco in eccesso restringe il lume delle vie aeree, provocando difficoltà respiratorie, violenti colpi di tosse e infezioni batteriche. Ne consegue un'infiammazione continua che lede il tessuto polmonare con possibile formazione di cicatrici e cisti.
Segni e sintomi a carico del sistema digerente
Alla presenza di muco sono legati vari problemi digestivi che possono diventare cronici. Alcuni bambini possono soffrire di ileo da meconio, un blocco intestinale che avviene poco dopo la nascita.
Molti problemi digestivi sono legati all'ostruzione del dotto pancreatico che riversa gli enzimi digestivi nell'intestino. I nutrienti non vengono assorbiti adeguatamente e ne conseguono diarrea o costipazione, malnutrizione, perdita di peso e ritardo nella crescita. In adolescenza anche la produzione di insulina può essere compromessa e dare luogo all'insorgenza di diabete mellito.
Complicazioni tipiche della fibrosi cistica a carico del sistema digerente sono pancreatite, responsabile di dolori molto forti, prolasso del retto, varie patologie epatiche, calcoli biliari.
Altri segni, sintomi e complicazione
Molti uomini affetti da fibrosi cistica soffrono di assenza congenita bilaterale dei vasi deferenti e sono pertanto sterili, ma possono ricorrere a dei trattamenti per la fertilità. Le donne possono portare a termine una gravidanza, tuttavia con serie problematiche nel corso della gestazione.
Nei malati di fibrosi cistica le ghiandole sudoripare tendono ad espellere un'eccessiva quantità di sali, provocando disidratazione, accelerazione della frequenza cardiaca e, quindi, affaticamento del cuore con possibile rischio di infarto, debolezza e bassa pressione sanguigna.
Altre complicazioni tipiche della fibrosi cistica sono il clubbing, ossia l'ingrossamento delle estremità delle dita di mani e piedi, e l'indebolimento delle ossa con conseguenti osteoporosi e osteopenia.
Cause genetiche della fibrosi cistica
La fibrosi cistica è una malattia autosomica recessiva dovuta a diverse mutazioni del gene CFTR, che codifica per un canale per il cloro presente sulle membrane degli epiteli. Il gene CFTR è localizzato sul braccio lungo del cromosoma 7.
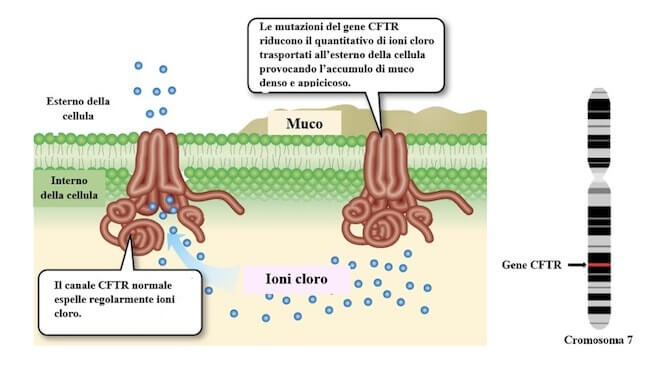
Il muco che ostruisce le cavità degli organi cavi nella fibrosi cistica è prodotto in seguito a disfunzioni del canale per il cloro codificato dal gene CFTR, localizzato sul braccio lungo del cromosoma 7.
Si conoscono sei classi di mutazioni responsabili delle manifestazioni fenotipiche della fibrosi cistica:
- le mutazioni di classe I sono responsabili della completa assenza della proteina CFTR;
- le mutazioni di classe II danno luogo a un'anomala localizzazione del canale;
- le mutazioni di classe III provocano alterazioni nel meccanismo di regolazione del canale che presenta un'attività limitata;
- le mutazioni di classe IV alterano la frequenza del flusso di ioni cloro;
- le mutazioni di classe V riducono il quantitativo di canali attivi;
- le mutazioni di classe VI provocano un turnover più alto delle proteine CFTR.
Le mutazioni delle classi I, II e III sono legate alle manifestazioni più gravi della fibrosi cistica.
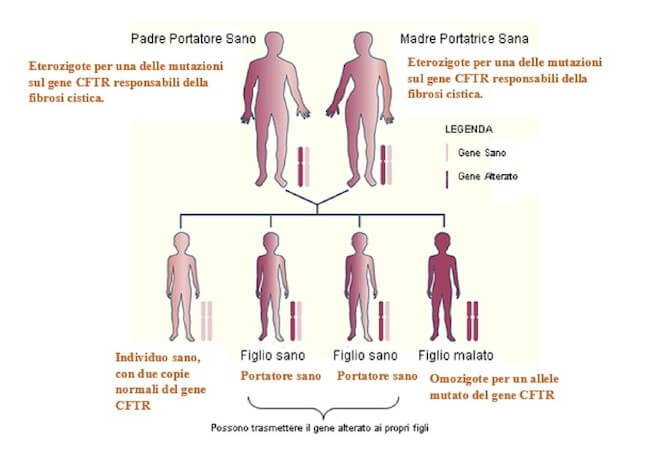
Le mutazioni responsabili della fibrosi cistica sono recessive. Un individuo malato è omozigote per un allele mutato del gene CFTR. Da due genitori eterozigoti o portatori sani può nascere un figlio affetto da fibrosi cistica con una probabilità del 25%.
Diagnosi e trattamento della fibrosi cistica
La fibrosi cistica può essere diagnosticata alla nascita con un test genetico o un'analisi al sangue. È anche possibile la diagnosi prenatale mediante amniocentesi o prelievo dei villi coriali.
Un tempo considerata fatale, le cure attuali consentono nei casi meno gravi il raggiungimento dei 40-50 anni di età.
Oltre al trattamento farmacologico sono raccomandati apposite terapie nutrizionali e respiratorie ed esercizio fisico. I pazienti sono inoltre inseriti in un programma di riabilitazione polmonare, ma nei casi più gravi si consiglia il trapianto del polmone.
Tra i farmaci maggiormente prescritti vi sono antibiotici, broncodilatatori e antinfiammatori.
L'esercizio fisico aiuta ad espellere il muco dalle vie respiratorie, ma per compensare l'eccessiva perdita di sali con la sudorazione, si raccomanda un'alimentazione più salata e l'uso di appositi integratori.
Per ovviare alla disfunzionalità digestiva si ricorre all'assunzione di enzimi digestivi e a supplementi di vitamine ed altri importanti nutrienti.
Link correlati:
Che cos'è la fenilchetonuria?
Che cos'è la corea di Huntington?
Studia con noi