Corea di Huntington
Che cos'è la corea di Huntington?
La corea di Huntington è una rara malattia neurodegenerativa a trasmissione autosomica dominante, caratterizzata dalla degenerazione di varie aree del sistema nervoso centrale, principalmente la regione dello striato.
Questa malattia è stata descritta per la prima volta in modo completo nel 1872 da George Huntington.
Sintomi
I sintomi tipici della malattia di Huntington sono corea, distonia, declino cognitivo e difficoltà comportamentali.
Nei portatori di un allele mutato del gene HTT, la corea di Huntington insorge tra i 30 e i 50 anni di età. Esiste anche una forma giovanile che compare intorno ai 20 anni.
Dalla comparsa dei primi sintomi la sopravvivenza media è di 15-20 anni; la principale causa di morte è la polmonite.
La prevalenza della corea di Huntington nella popolazione caucasica è di 5-10 ogni 100.000. In Giappone, Cina e Finlandia si registra una prevalenza circa dieci volte inferiore.
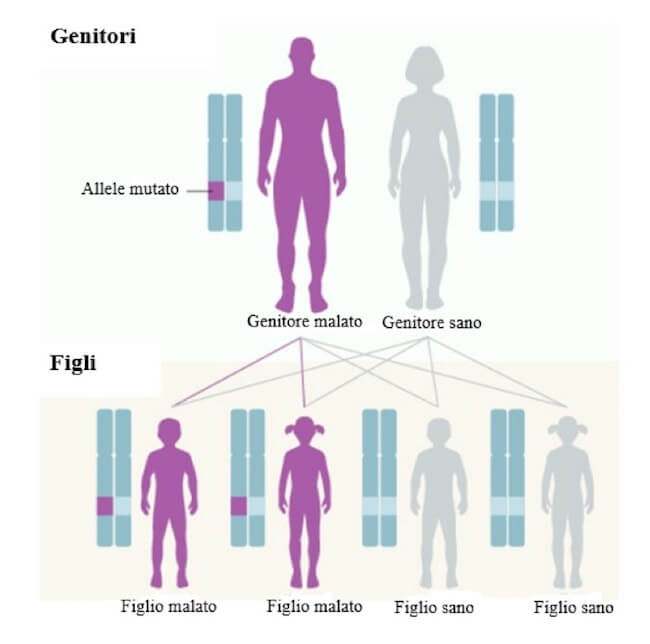
La corea di Huntington è una rara malattia autosomica dominante. Un genitore affetto (eterozigote per una mutazione dominante in un gene specifico) ha la probabilità del 50% di avere figli (indipendentemente dal sesso) portatori della stessa mutazione e che manifesteranno in età adulta i sintomi della corea di Huntington.
Cause genetiche
La causa della corea di Huntington è un numero elevato di ripetizioni della tripletta CAG nell'esone 1 del gene HTT, localizzato sul braccio corto del cromosoma 4 e che codifica per una proteina chiamata huntingtina.
La tripletta CAG codifica per l'aminoacido glutammina e, negli individui normali, è ripetuta tra 9 e 26 volte.
Gli alleli con 29-35 ripetizioni sono detti intermedi in quanto non sono direttamente responsabili della malattia, ma inducono frequentemente errori durante la duplicazione del DNA, incrementando il grado di espansione della tripletta CAG nel gene delle nuove copie di DNA.
Se tale errore interessa le cellule della linea germinale è probabile la nascita di un figlio che rechi un allele associato alla malattia.
Le mutazioni del gene HTT associate alla corea di Huntington
Gli alleli del gene HTT determinanti la comparsa della corea di Huntington hanno un numero di ripetizioni superiore a 36.
Da 36 a 39 ripetizioni la penetranza della malattia non è completa e l'insorgenza può avvenire tardivamente, mentre in presenza di 40 ripetizioni e più la penetranza della malattia è completa e sintomi tendono a insorgere tanto più precocemente quanto più alto è il numero di ripetizioni.
La forma giovanile della corea di Huntington è provocata da un'espansione superiore a 55 volte della tripletta CAG.
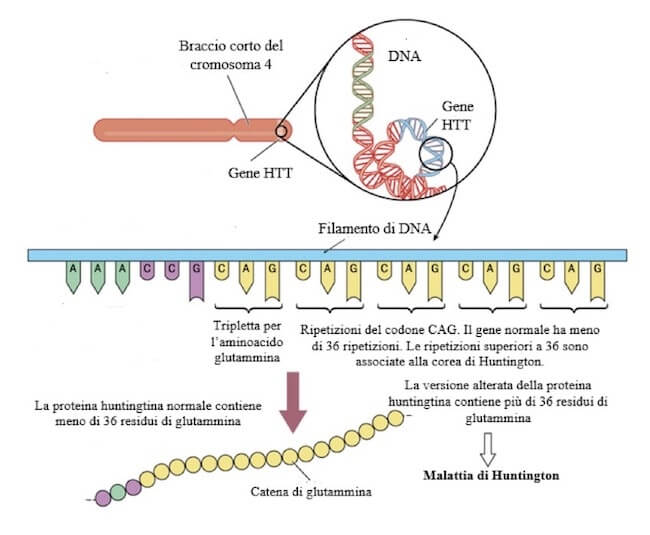
La causa genetica della corea di Huntington è l'espansione delle ripetizioni della tripletta CAG nel gene HTT, localizzato sul braccio corto del cromosoma 4. La tripletta CAG codifica per l'aminoacido glutammina. Ripetizioni superiori a 36 del codone CAG provocano la sintesi di versioni anomale della proteina huntingtina, contenenti più di 36 residui di glutammina.
Ruolo della proteina huntingtina
Il ruolo della proteina huntingtina non è ancora chiaro, ma sembra essere coinvolta nella funzionalità sinaptica e nello sviluppo del sistema nervoso prima e dopo la nascita, svolgendo un'azione anti-apoptotica.
La proteina huntingtina è espressa in molti tessuti, ma i livelli più alti si riscontrano nel cervello.
La distruzione e la perdita di funzionalità di molte aree del cervello sono state legate in alcuni casi alla presenza di inclusioni citoplasmatiche e nucleari di frammenti derivati dalla rottura delle versioni molto lunghe della proteina huntingtina.
Tuttavia, non è noto se tali inclusioni siano la causa alla base dei meccanismi patologici della corea di Huntington o dei prodotti secondari di altri meccanismi.
Sintomi e segni clinici della corea di Huntington
La corea di Huntington è così chiamata perché caratterizzata, soprattutto nella fase iniziale, da movimenti incontrollabili e involontari.
Questi movimenti interessano inizialmente le estremità del corpo, come le dita di mani e piedi, e i muscoli facciali, per poi estendersi al resto del corpo.
Col passare del tempo, il movimento e il mantenimento dell'equilibrio vengono compromessi e svolgere le normali attività quotidiane, compresi mangiare e bere, diventa sempre più difficile.
Negli stadi avanzati della malattia si osservano ipocinesia, acinesia e rigidità del corpo, ma la corea a tratti persiste.
La progressione della malattia è accompagnata da declino cognitivo e da vari disturbi psichici, soprattutto ansia, depressione, apatia, irritabilità e atteggiamenti ossessivo-compulsivi.
Il quadro clinico completo è paragonabile a quello della schizofrenia e spiega l'elevata tendenza al suicidio sin dagli esordi della malattia.
Altre manifestazioni cliniche della corea di Huntington sono la perdita di peso e l'alterazione dei ritmi sonno-veglia, a causa della perdita di neuroni nelle aree nervose interessate, come l'ipotalamo.
Diagnosi e trattamento
La diagnosi della corea di Huntington si basa sulle tipiche manifestazioni cliniche in pazienti con un genitore colpito dalla stessa malattia, anche se il test del DNA per la verifica del numero di ripetizioni della tripletta CAG nel gene HTT rappresenta ormai la procedura più accurata e affidabile.
Non esiste una terapia specifica per la corea di Huntington, in quanto potenziali farmaci sono ancora in fase di studio e sperimentazione. Il trattamento consiste pertanto in specifici interventi per migliorare la qualità della vita e limitare la sofferenza dei pazienti.
Anticipazione nella trasmissione della corea di Huntington
Nella trasmissione da una generazione all'altra della malattia di Huntington si osserva il fenomeno dell'anticipazione, in conseguenza del quale la comparsa dei sintomi avviene prima che nel genitore e le manifestazioni cliniche sono spesso più gravi.
L'anticipazione avviene in misura di gran lunga maggiore per via paterna, a causa dell'elevata instabilità della tripletta CAG durante la spermatogenesi e il conseguente aumento del numero di ripetizioni.
Atropina e reserpina sono esempi di farmaci impiegati da pazienti affetti da tale malattia.
Link correlati:
Che cos'è e a cosa è dovuta l'anemia falciforme?
Che cos'è la sindrome di Klinefelter?
Che cos'è la sindrome di Laron?
Studia con noi