Osteogenesi imperfetta
Che cos'è l'osteogenesi imperfetta?
L'osteogenesi imperfetta, nota anche come "malattia delle ossa fragili", è un gruppo di disordini genetici che impediscono la corretta formazione delle ossa, causandone debolezza, deformità e tendenza alla rottura, anche in seguito a traumi lievi o a nessuna ragione specifica.
La causa di questa malattia è un'alterazione della struttura e dell'assemblaggio delle fibre collagene, presenti in tutti i tutti i tessuti connettivi.
Oltre a debolezza delle ossa, l'osteogenesi imperfetta si caratterizza per fragilità dei denti, debolezza dei muscoli e perdita dei capelli.
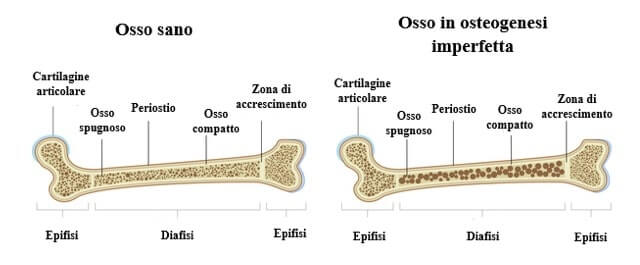
Immagini a confronto di osso sano e osso in caso di osteogenesi imperfetta.
Tipi di osteogenesi imperfetta
Sono state descritte otto forme di osteogenesi imperfetta, indicate con i numeri romani da I a VIII.
La forma più lieve è il tipo I, quella più grave il tipo II. Le altre forme vengono distinte sulla base dei segni e sintomi, la cui gravità è distribuita tra il tipo I e il tipo II.
Spesso le caratteristiche mediche delle diverse forme di osteogenesi imperfetta si sovrappongono. L'analisi genetica può contribuire alla diagnosi.
Gli individui affetti da osteogenesi imperfetta di tipo I manifestano rottura delle ossa durante l'infanzia e l'adolescenza. Da adulti le rotture sono meno frequenti, ma molti individui perdono i capelli. Altra caratteristica dell'osteogenesi di tipo I è la tendenza della sclera degli occhi ad assumere una tonalità tra l'azzurro e il grigio.
Nelle altre forme di osteogenesi imperfetta tra gli altri sintomi si hanno bassa statura, problemi respiratori e difetti nella formazione dei denti.
Il tipo II si caratterizza per fragilità della gabbia toracica e polmoni non sviluppati correttamente. I bambini spesso muoiono pochi giorni dopo la nascita.
L'osteogenesi imperfetta colpisce tra i 6 e i 7 individui ogni 100.000 in tutto il mondo. I tipi I e IV sono le forme più diffuse, con un'incidenza di 4-5 individui ogni 100.000.
Cause genetiche dell'osteogenesi imperfetta
L'osteogenesi imperfetta è dovuta a mutazioni nei geni COL1A1, COL1A2, CRTAP e P3H1.
Le mutazioni nei geni COL1A1 e COL1A2, che codificano per il collagene di tipo I, provocano circa il 90% dei casi di osteogenesi imperfetta, principalmente i tipi II, III e IV.
L'osteogenesi imperfetta di tipo I è, nella maggior parte dei casi, provocata da sole mutazioni nel gene COL1A1.
Le mutazioni nei geni COL1A1 e COL1A2 hanno quasi sempre un'ereditarietà di tipo autosomico dominante. Mutazioni recessive sono state descritte per alcuni casi di osteogenesi imperfetta di tipo III.
Le mutazioni nei geni CRTAP e P3H1 causano forme rare ma gravi di osteogenesi imperfetta, principalmente i tipi VII e VIII. Questi geni codificano per proteine che contribuiscono alla maturazione e al corretto assemblaggio delle fibre collagene nel tessuto connettivo. L'ereditarietà delle mutazioni è di tipo autosomico recessivo.
Le forme V e VI di osteogenesi imperfetta dipendono da mutazioni in geni non ancora identificati.
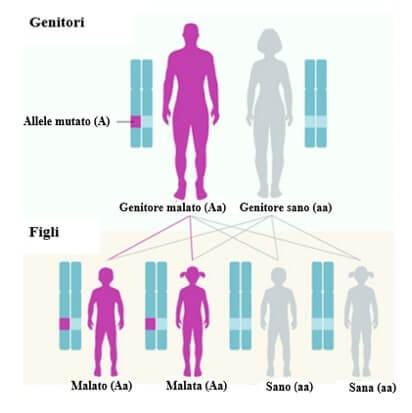
In caso di ereditarietà autosomica dominante basta un allele mutato perché la malattia si manifesti. Da un genitore malato (Aa) e uno sano (aa) vi è il 50% di probabilità che nascono figli malati. Le mutazioni nei geni COL1A1 e COL1A2, responsabili della maggior parte delle forme di osteogenesi imperfetta, si trasmettono in questo modo.
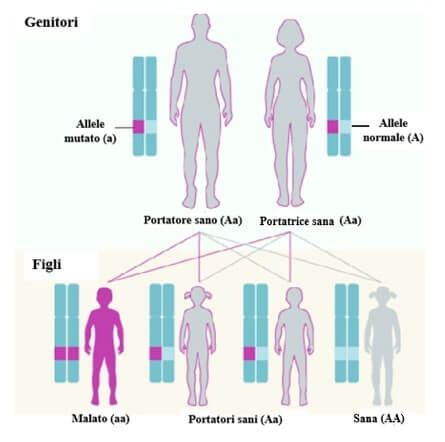
In caso di ereditarietà autosomica recessiva la malattia si manifesta se entrambi gli alleli di un dato gene sono mutati. Da due genitori normali, ma portatori sani (eterozigoti Aa), vi è il 25% di probabilità che nasca un figlio malato (aa). Le mutazioni nei geni CRTAP e P3H1 e alcune nei geni COL1A1 e COL1A2 si trasmettono in questo modo.
Link correlati:
Che cos'è e a cosa è dovuta l'anemia falciforme?
Che cos'è la sindrome di Laron?
Che cos'è il nanismo ipofisario?
Studia con noi